
Is Insulin Resistance Really Making Us Fat?
Is Insulin Resistance Really Making Us Fat?

Disclaimer: I love and respect many low-carbers and low-carb researchers, and think low-carb diets are very appropriate and perhaps necessary for many people.
Many in the low-carb field seem to think that insulin resistance is what is making us fat and even that insulin resistance is caused by… insulin. Too much carb makes too much insulin and the insulin receptors get scared and run away. Intriguing theory, but very simplistic. During the Q&A session of my Wise Traditions talk this weekend, I made the following quip:
Saying that insulin causes insulin resistance is like saying that childhood mortality is caused by children.
Melissa McEwen caught this quote on her live twitter feed. Melissa also live tweeted many other excellent talks from the conference, including Stephan Guyenet's masterpiece on the traditional diet of the Pacific islands. He uncovered islands where the traditional diet was over 90% carbohydrate and other islands where the traditional diet was mostly fat, including a whopping 50% of calories just from saturated fat alone. Neither of the populations had insulin resistance, diabetes, or cardiovascular disease, and none of either island's inhabitants were fat. In came refined foods, and they became vulnerable to all of these diseases.
Lots of other people live tweeted talks and other events from the conference, and you can see a hodge podge of these tweets here.
In this blog post, I'd like to review a couple animal models that strongly suggest that between insulin resistance and leptin resistance, leptin resistance is much more critical to the development of obesity.
The mainstream often cries “eat less and exercise more” whenever someone has trouble losing weight. Well in fact there's a little molecule called leptin that our fat tissue makes, which slips into our brain, acts on the hypothalamus, and makes us… *drumroll*… eat less and exercise more. Well, at a minimum it makes us eat less and expend more energy when we exercise.
Even though leptin appears to decrease food intake and increase energy expenditure, obese people have very high levels of leptin compared to lean people (1). When they lose weight, leptin levels go down, but stay way above what you'd find in a lean person. The drop in leptin is accompanied by a drop in thyroid hormone (2, 3), muscular utilization of glucose (3), sympathetic nervous system activity (3), and adrenaline (3), and by changes in the pattern of brain responses to food (4), all of which are reversed by injections of leptin. These studies have been very small and preliminary, but they support the widely held belief that obese people are leptin resistant.
Insulin has a century or so more research behind it than leptin has, so insulin resistance is a much more well defined phenomenon than leptin resistance. Recent data suggest that about 39% of overweight Americans have either diabetes, impaired fasting glucose, or elevated fasting insulin (5), which are the clinical manifestations of insulin resistance.
These numbers are pretty similar to what Zelman reported in 1952 (6). Zelman wrote long before the obesity epidemic emerged and it took him 18 months, a full year and a half, to find 20 obese people who weren't alcoholics. He reported that about half of these people were glucose intolerant, meaning that when they were given a load of glucose, their insulin couldn't work fast enough to prevent an abnormal spike in blood sugar.
Zelman's finding was similar to what had already been reported in larger groups. His new contribution was to show that upon liver biopsy, all patient but one — a full 95% — showed signs of at least mild to moderate liver damage. The longer the people had been obese, the more damaged their liver was. Long before the discovery of leptin, a hormone that acts on the hypothalamus, Zelman hypothesized that damage to the hypothalamus caused obesity and cravings for nutrient-poor sweets and fats, that the consumption of too much sugar and fat without sufficient choline, protein, and other nutrients led to liver damage, and he cited another researcher's suggestion that liver damage led to glucose intolerance.
Why would liver damage lead to glucose intolerance? The liver not only contributes to clearance of glucose from blood, but, more importantly, the liver produces glucose from protein in a process called gluconeogenesis. Ordinarily, the liver stops making glucose in response to insulin. However, if liver damage prevents this response, the liver will keep making glucose even when we don't need any more of it. More glucose in the blood will cause the pancreas to make more insulin, but the insulin will fail to stop the liver from making more glucose, and a vicious circle will ensue. With increasing levels of glucose and insulin in the blood, many other tissues such as skeletal muscle and fat may deliberately stop responding to insulin themselves in order to prevent glucose overload in their own cells.
The fact that insulin resistance is not found in all obese people does not mean it plays no causal role, because obesity does not necessarily have the same cause in every person.
Nevertheless, a look at the genetic animal models of leptin and insulin resistance would suggest that leptin resistance has a much more prominent role in causing obesity and that insulin resistance without leptin resistance may not cause obesity at all.
The agouti yellow mouse was the first genetic animal model of obesity, systematically described as far back as the 1920s, and it later turned out to be leptin resistant (7). Another early model of obesity was neither dietary nor genetic, but rather involved the removal of the hypothalamus, the principal (but not only) site of leptin action (8). The common modern genetic models of obesity include the ob/ob mouse (9), the db/db mouse (10), and the fa/fa rat (10). The ob/ob mouse doesn't produce leptin at all, while the db/db mouse and the fa/fa rat have defects in the leptin receptor. All three types of animals become insulin resistant and fat.
It's a little bit harder to study the effects of genetic insulin resistance, because mice with no insulin receptors die 2-3 days after birth (11). Nevertheless, mice can be developed with deletions of the insulin receptor in specific tissues. Deletion of the insulin receptor from liver tissue results in whole-body (systemic) insulin resistance (12). As predicted above, the insulin resistance in the liver can cause systemic insulin resistance by causing the liver to continue making glucose and sending it out into the blood stream even when it isn't needed.
But, surprise surprise! These mice become neither leptin resistant nor fat (13). In fact, while the effect is not statistically significant, they are slightly more sensitive to leptin and slightly more lean.
Although glucose metabolism tends to normalize after six months, fasting glucose is initially higher. Feeding them glucose produces much higher blood glucose peaks and dosing them with insulin produces much less effective declines in blood glucose. What is particularly remarkable, however, is their high insulin levels. These dramatic changes persist even through adulthood.
In the fasting state, insulin levels are over 7-fold higher:
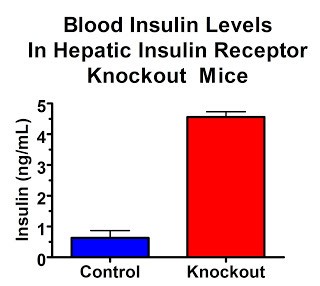
In the fed state, insulin levels are 23-fold higher:
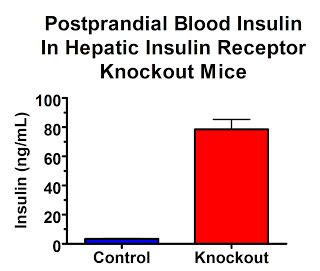
Their insulin receptors are only knocked out in their liver. So if high insulin levels are what act on our adipose tissue to make us fat, these mice should be really, really, really fat.
On the contrary, they are quite lean:
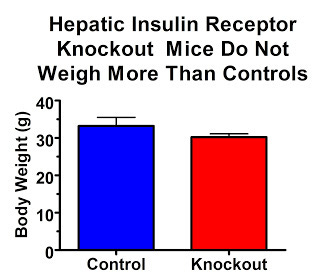

Why aren't they fat? This study showed that they were just as sensitive to leptin, perhaps slightly more sensitive, than controls. Thus, while leptin resistance and insulin resistance often go together, it seems that leptin resistance is a much more important contributor to obesity.
These data should not be considered evidence that insulin resistance can never lead to leptin resistance in humans. In fact, human hepatic insulin resistance (insulin resistance of the liver) looks nothing like what happens in the hepatic insulin receptor knockout mice. The livers of these mice don't respond to insulin at all. In humans, this insulin resistance is “selective.” The livers of “insulin resistant” humans continue to manufacture fat and send it out into the blood as triglycerides in response to insulin, but fail to suppress the production and export of glucose in response to insulin.
In humans, insulin resistance of the liver leads to increased triglycerides in the blood. One theory that has some experimental support, but is still questioned by some experts, is that increased blood triglycerides decrease the transport of leptin into the brain. For this and perhaps other reasons, insulin resistance as it occurs in humans could, perhaps, cause leptin resistance. However, if it does not cause leptin resistance, it is very unlikely to make people fat.
In describing this selectivity, Dr. Robert Lustig recently made the following remark (14):
The reason for this uncoupling of insulin's two main hepatic signaling pathways remains unclear.
I propose that the explanation may be rather simple. Rather than a result of gluocose toxicity or fat toxicity or fructose toxicity, the development of insulin resistance may be a natural, protective, homeostatic response to energy overload. It certainly has adverse consequences down the line, but the selectivity does seem to suggest a deliberate attempt of the liver to alter its energy metabolism. Moreover, the liver is clearly modifying its energy metabolism in a consistent way that exports energy. In this case, it is exporting both glucose and fat at the same time, rather than suppressing one whenever it engages in the other.
I'll expand on these ideas in an upcoming post arguing that the main culprit preventing the liver from correctly handling its energy is suboptimal intakes of choline, and perhaps lipid peroxidation, which prevent the liver from being able to export the fat that it obtains from dietary fat or that in manufactures anew from things like fructose and ethanol. Nutrient deficiencies and other issues that compromise the liver's ability to burn energy are also likely involved.
First, expect a brief review debunking some of the conventional ideas about the so-called “Receptor for AGEs (RAGE).”
Then we can go back to fruit and honey.