
Where Do Most AGEs Come From? O Glycation, How Thy Name Hast Deceived Me!
Where Do Most AGEs Come From? O Glycation, How Thy Name Hast Deceived Me!

I've written a few posts about advanced glycation endproducts (AGEs) in the past, which can be found here. These posts include a refutation of the common belief that the “receptor for AGEs” (RAGE) is actually a receptor for AGEs, and a refutation of the implausible and unreliable data suggesting that butter is a major source of dietary AGEs. People have recently been asking me to write more about AGEs (see here and here), especially about the role of high blood sugar in promoting the formation of these compounds and thereby contributing to cellular dysfunction and disease.
There are a lot of misconceptions about AGEs, and one of them is that they are mostly formed from glucose directly glomming on to proteins. The term “glycation,” which is clearly derived from “glucose,” certainly contributes to this misconception, but the situation is actually much more complex than this. Glucose does indeed have the hots for proteins, but the high school glycation prom has a sexy chaperone named fructosamine 3-kinase who's kicking carbonyls and taking names, and if the two dance too close, F3-K steps in the way.
It is instead the sneaky dicarbonyls (pronounced like “DIE-carb-o-NEELS”) that escape the attention of our otherwise striking chaperone. They are on average 20,000 times more reactive than glucose, and they emerge from the broken pieces of glucose, protein, and fat — and not just PUFAs. Nevertheless, they do no harm unless they slip past our good friend glutathione, who polices the streets at night and renders the balance of these creepy would-be criminals as impotent as the mythical sorcerer lurking in the shadows of Maasai-land.
In future posts, I will explain why I believe that AGEs formed within our bodies do indeed contribute to disease but are nevertheless likely to emerge as essential components of cellular communication, and why they add further colors to the portrait being painted in which disease is seen as communication gone wrong. In this post, I will simply attempt to explain what we do and don't know about where the AGEs within our bodies come from.
The Maillard Reaction
Humans have presumably known since the dawn of ancient campfires that cooking causes food to brown and develop unique flavors. It wasn't until the emergence of the twentieth century, however, that the French physician and chemist Louis Camille Maillard produced the first detailed outline of the chemical reactions involved and the implications of the process for many branches of modern science. We refer to this process as “The Maillard reaction” in his memory. The late 1960s hailed the first discovery that a similar reaction occurs in the blood of diabetics, producing HbA1c, a form of hemoglobin that's been damaged by glucose. Scientists then coined the term “glycation” to distinguish this aberrant and apparently pathological process from the normal and necessary “glycosylation” reactions that our enzymes deliberately carry out (1).
Although the division is somewhat artificial, we generally split the Mallaird reaction up into three stages: early, intermediate, and late (or “advanced”). HbA1c is an example of an “early glycation” product. It may seem strange, but because glucose flip-flops around a bit when it reacts with hemoglobin or other proteins during early glycation, it winds up producing messed up amino acids with names like “fructosyl-lysine” and “fructosyl-valine.” Early glycation products derived from fructose have different names altogether. The “intermediate glycation” products are the dicarbonyls, and it is the dicarbonyls that produce most of the AGEs. But here's the catch: dicarbonyls can be formed in a number of ways, including some that have nothing to do with glucose. They can even be formed enzymatically during fermentation, and in fact they are an active antibacterial ingredient of manuka honey (1).
In case you missed Mat Lalonde condensing two semesters of organic chemistry into 45 minutes at the Ancestral Health Symposium, here's what a dicarbonyl looks like:
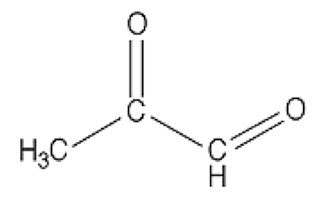
This bad boy right here is named methylglyoxal. You can see there are two carbons (C) that are double-bonded to oxygen atoms (O). Each of these C=O groups is called a “carbonyl.” Because there are two of them, methylglyoxal is called a “dicarbonyl.”
If there are any dads in the audience, you should feel your protective instincts kicking in now, because whenever you see two carbonyls paired up like this you can bet your bottom dollar someone's about to crash the glycation prom while the F3-K chaperone's fixing her hair in the bathroom. And besides, you can tell just by the way his unkempt carbonyls are flailing about that he's probably never held a job down for more than three months. A piece of advice: if methylglyoxal comes home for dinner, as a friendly gesture you may want to introduce him to your gun collection.
There are quite a number of other dicarbonyls, but as we'll see below methylglyoxal's primary partners in crime are glyoxal and 3-deoxyglucosone. Each of these dicarbonyls leaves a “fingerprint” of sorts by producing a different array of AGEs. Thus, if we know a little about where these dicarbonyls come from, we can attempt to solve the “Who Dunnit” by seeing which AGEs we find on the scene and thus tracing where they came from.
Before we begin to solve the crime, I should note that I will be giving preference to studies using chromatographic techniques to measure AGEs. For more details, see here.
Dietary Sources of AGEs
The available evidence suggests that we do absorb certain AGEs from the intestinal tract, but almost immediately begin peeing them out. As a result, dietary AGEs are unlikely to contribute meaningfully to the pool of AGEs circulating in our bodies at any given moment.
The most common AGEs in foods that have been measured so far are pyrraline and methylglyoxal-derived hydroimidazolone-1 (1). Yikes! Try to say that three times fast. To make it a little easier, we'll call that latter compound MG-H1.
The authors of a 2005 study lasting nine days (2) divided eighteen people into four groups to estimate to what degree dietary pyrraline could be absorbed and excreted in the urine. One group served as a control and the other three consumed meals based on either pretzel sticks, coffee brew, or custard, each containing known amounts of pyralline. The authors concluded that somewhere between 40 and 65 percent of dietary pyrraline winds up in the urine, and that dietary pyralline accounts for 90 percent of the pyralline found therein. These results strongly suggest that dietary pyralline is absorbed, but we cannot rule out with complete certainty the possibility that certain foods increase the amount of pyralline we make within our own bodies. On the other hand, assuming it is absorbed, we do not know to what extent some of it may stay within our bodies where it could, at least hypothetically, do some damage.
This group recently published several studies using tissue cultures that mimic the human intestinal lining providing further evidence for the absorption of pyralline and some other AGEs (3, 4). These studies suggested that pyrraline is absorbed when it is present as a dipeptide (two amino acids linked together), but once the cell takes up the dipeptide it splits it in half and releases free pyrraline into the bloodstream. The group also studied its interaction with kidney cells and concluded that pyrraline is likely to be rapidly excreted into the urine. When they previously looked for pyrraline in the blood of people who had been eating their usual diets, they couldn't find any (5).
Although we can't rule out with complete certainty the possibility that some pyrraline is absorbed and decomposes into some other compound that hangs around in our bodies, experiments with laboratory animals show that even when we inject the animals with radioactively labeled AGEs, almost all of the radioactive label disappears into the urine within two hours (6). The most logical conclusion at the moment is that pyrraline absorbed from food just doesn't hang around in the body for very long.

The same tissue culture studies that suggested pyrraline makes its way from the gut into the bloodstream also suggested that our intestinal cells absorb a small amount of MG-H1 and tightly hang on to it. Generally these cells last less than a week before they slough off and get taken out the other end with the trash, so these experiments would suggest that MG-H1 is excreted in the feces rather than absorbed.
When we consider that pyrraline is absorbed from food but not present in the bloodstream while MG-H1 appears not to be absorbed from food but is nevertheless abundant in the bloodstream (7), and when we consider the evidence below that as long as our kidneys are working we quickly pee out most free AGEs regardless of where they come from, we have to conclude at least for the time being that most AGEs within our body at any given moment do not come from food.
The Scene of the Crime: The Cell
Few people are better poised to begin marking the crime scene than PJ Thornalley. Truly the Gil Grissom of AGEs, Thornalley has an automated robotic column-switching system that allows him to quantify nineteen different markers of glycation, oxidation, and nitration all at once using the “gold standard” technology of stable isotopic dilution analysis with liquid chromatography and tandem mass spectrometry (8).
Thornalley's group has provided some interesting evidence suggesting that most AGEs occur inside our cells, not in our blood plasma. Here are the results for several healthy volunteers, showing much higher concentrations of AGEs in blood cells than in plasma (7):

In the same paper, this group showed that the protein-adjusted concentration of AGEs in the plasma of rats is quite similar to that of skeletal muscle and nervous tissue. Nevertheless, when we consider that these values represent the likelihood of a particular amino acid within a protein being attacked by an AGE, and that almost 90 percent of our proteins are contained within our cells, it would seem pretty likely that most AGEs occur inside rather than outside our cells.
I should take a moment to acknowledge that certain AGEs accumulate with age on long-lived extracellular proteins like skin collagen (9), and as a result it is possible that as we grow older the proportion of AGEs existing outside our cells might increase. There is not enough data yet to say for sure. Nevertheless, we will see below that even in the cases where the scene of the crime is outside the cell, it is the cell that acts as a breeding ground for the criminals, those bad boys we like to call the dicarbonyls.
We Actively Degrade AGE-Modified Proteins and Flush the AGEs Away
Our cells have a number of different ways by which they recognize damaged proteins and degrade them, thereby both protecting themselves and providing themselves with a fresh batch of amino acids to start over and build the needed proteins anew. Glycated proteins are at a minimimum subjected to the normal robust turnover of cellular proteins, and may be subjected to these more rigorous forms of clearance as well (10).
Thornalley's data (7) suggest that we are constantly degrading AGE-modified proteins down to their constituent amino acids, liberating the free AGEs, and flushing them down the drain. Free AGEs are found at extremely low levels in plasma, constituting less than two percent of all AGEs measured therein. Far be it from us to believe that this is because most AGEs that traverse the bloodstream are not free! Nay, let us not persecute these AGEs with such oppressive thoughts. It is, my friends, simply because these liberated AGEs have swum to sea, basking in the brilliant yellow waters that shall set them free:
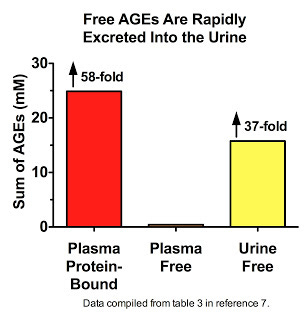
His data also show that in patients with kidney disease, free AGEs accumulate in plasma but are cleared by dialysis (11).
If we simply consider the fact that we pee out some one or two liters of water per day but have barely more than three liters of water for plasma volume, it would certainly seem that AGEs are going nearly as often as they're coming. We can't use these data to trace the daily history of AGE formation and AGE liberation, but we can imagine that the flux is just massive.
Dusting For Fingerprints — Who Dunnit?
If we want to solve the crime and determine just who put their hands on what and when, we need only dust for fingerprints. Sugars can generate AGEs through a stepwise process during the Maillard reaction, but these AGEs target lysine and the reaction is rather slow, whereas dicarbonyls target both lysine and arginine and the reaction is nimble and quick (12). Since arginine-AGEs outnumber lysine-AGEs within the body by 10-100-fold (7), we can be quite confident that it is mostly dicarbonyls doing the damage. Each dicarbonyl generates a specific array of AGEs, so by determining which AGEs are most abundant in our bodies, we can identify the guilty parties. This can help us figure out what dietary factors and metabolic processes are most likely to promote AGE formation. Here's what these fingerprints look like (7):
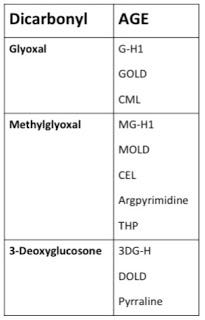
There are actually a lot of other dicarbonyls that can form AGEs, but these three are by far and away the most prominent of the bunch (12). Among the Three Glycateers, who's doing the most damage? Let's take a look, again using data from Thornalley's group.
Here we see that most AGEs in plasma are derived from methylglyoxal and 3-deoxyglucosone (13), and that it is methylglyoxal-derived AGEs that increase the most in diabetes:
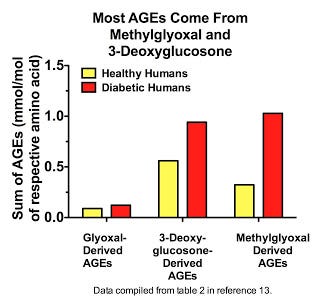
Similar results were seen within the blood cells of healthy human volunteers, although 3-deoxyglucosone was more prominent than methylglyoxal in these cells (7). Similar results were also seen in rats for a wider range of tissues, except that the increases seen with diabetes all the more overwhelmingly resulted from methylglyoxal-derived AGEs (7). By far and away, MG-H1 and 3DG-H are the most prominent AGEs. Thornalley's group had previously shown that MG-H1 and its close cousin MG-H2 are the major AGEs in human lens protein (14), although they didn't measure 3DG-H. Thus, most AGEs within the human body appear to be formed from 3-deoxyglucosone and methylglyoxal.
AGEs Come From Carbohydrate, Protein, and Fat
Our task now is quite simple. We know who's crashing the party, but why? Where are all these dicarbonyls coming from?
Light-induced oxidation of polyunsaturated fatty acids (PUFAs) produces glyoxal and methylglyoxal, but glyoxal is much more prominent (15). Iron-induced oxidation of PUFAs also produces glyoxal (16). Copper-induced oxidation of the PUFAs in the LDL membrane, or simple incubation of PUFAs with protein, will produce carboxymethyl-lysine (CML), a glyoxal-derived AGE (17). Nevertheless, as described above, glyoxal-derived AGEs make only a minor contribution to the AGEs found within our bodies. Even the data above are likely to greatly exaggerate the role of glyoxal, because AGEs derived from glyoxal are almost seven times more stable than those derived from methylglyoxal (7). We must therefore reach the verdict that peroxidation of PUFAs is very unlikely to be a major source of AGEs.
Are PUFAs off the hook? Not at all. We will see below that oxidative stress is a central factor in AGE formation, and guzzling corn oil gets that oxidative stress a-goin'. I'll return to this topic in future posts.
The spontaneous degradation of carbohydrates can produce dicarbonyls, but this is generally a very slow process that requires high concentrations of sugars, heat, and bases such as phosphate ions in order to accelerate meaningfully, and thus mainly becomes significant in the heat-processing of foods (12).
Nevertheless, when sugars glom on to proteins they do indirectly lead to meaningful amounts of AGEs. That sexy chaperone and deglycation enzyme F-3K uses ATP, a major currency of cellular energy, to separate the two (18). For example, glucose will react with a protein to produce a fructosamine; F-3K will step in the way, releasing the protein from glucose's grip. But glucose walks away angry, not simply because he's lost his carnal love, but because F-3K actually had the nerve to turn him into a fructose 3-phosphate! In his anger he implodes, morphing spontaneously into 3-deoxyglucosone and ready for the attack. As we've seen, 3-deoxyglucosone is a major contributor to the AGEs present within our bodies. We must therefore reach the verdict that, in this indirect way, glucose is indeed causing some trouble.
Since deglycation occurs within the cell, moreover, this is further evidence that the cell is a breeding ground for these dangerous dicarbonyls.
The data we reviewed above, however, suggest that methylglyoxal accounts for the majority of the increased AGEs seen in diabetes, which may indicate that methylglyoxal's importance grows as a situation becomes pathological. There is thus more to the story than glucose: methlglyoxal, in fact, comes from carbohydrate, protein, and fat.
When glucose or fructose enter glycolysis, about 0.1-0.4 percent of these molecules will slip and fall off one of the enzymes, and spontaneously convert into methylglyoxal (19).
We possess enzymes that usually convert the amino acid threonine to glycine and acetyl CoA. Glycine is another important amino acid and acetyl CoA is just a carrier for acetate, which is a two-carbon, energy-rich compound and the same acetate you'd find in vinegar. When we're producing more acetyl CoA than we can burn, however, this reaction gets backed up and these enzymes instead convert threonine to aminoacetone and then to methylglyoxal. Some authors have suggested that this pathway is likely to become meaningful during caloric restriction, ketogenic dieting, and diabetes (20, 21), but no one has clearly quantified how important it becomes.
When we burn fats for energy, our adipose tissue releases free fatty acids into the bloodstream and our liver converts them into ketones, sending the ketones back into the blood. Once there, some of these ketones will generate acetone, which is responsible for “ketone breath.” Our cells will then use an enzyme called CYP2E1 to convert the acetone to methylglyoxal (22, 23, 24). This is the same enzyme that contributes to the toxicity of Tylenol poisoning and chronic alcohol abuse. Basic logic would suggest that this pathway would also become more important during caloric restriction, ketogenic dieting, and diabetes, because these conditions increase the production of ketones.
Exactly how important these different ways of producing methylglyoxal are is still up in the air, but there are a couple of studies suggesting that ketones and perhaps threonine become important sources in situations where free fatty acids increase:
A 2005 study (25) showed that a group of subjects told to read Dr. Atkins' New Diet Revolution and follow the diet experienced a nearly 70 percent increase in methylglyoxal levels along with a six-fold increase in acetone. The subset of subjects with evidence of ketosis and successful weight loss experienced a doubling of methylglyoxal concentrations. This study is difficult to interpret because there was no control group. The authors, moreover, did not give any dietary counseling to the subjects or stress the importance of consuming liver and leafy greens in order to obtain sufficient B vitamins, lipoic acid, and other nutrients needed to metabolize energy efficiently.
A study published last year (26) showed that citrate reduced the formation of a methylglyoxal-derived AGE in the lens protein of diabetic rats. Although it obliterated the massive increase in ketone levels seen in diabetes, it had no effect on hyperglycemia.
Regardless of how important these different pathways are, it would be quite silly to blame AGEs on “carbohydrate,” or “protein,” or “fat,” because these dicarbonyls cause nary a whiff of harm unless they slip past our good friend glutathione.
Zinc, Insulin, and Glutathione: The Dicarbonyl Defense Team
Our cells possess two zinc-dependent enzymes, glyoxalase-1 and -2, that detoxify dicarbonyls in a two-step process (19). In the case of methyglyoxal, they convert it to D-lactate, which can be used for energy or as a source of glucose. But it's that cape-wearing, free radical-wrestling, toxicant-thwarting tripeptide named glutathione that glues together this dazzling detoxification defense team. He quickly tackles any of the dangerous dicarbonyls in sight, pinning them down, rolling them out, bending and twisting them into all sorts of painful, pretzel-like permutations until he's finally formed them into a hemithioacetal. The two glyoxalase enzymes are then able to incarcerate the hemithioacetal for a brief bit, rehabilitate the dicarbonyl, and free the final product, letting glutathione go so he can continue fighting the good fight.
Treatments that deplete cells (27) or live animals (28) of their glutathione cause large increases in methylglyoxal concentrations, suggesting that the glyoxalase system is ordinarily efficiently detoxifying much or most of the methylglyoxal that crosses its path. Little is known about the molecular mechanisms by which our cells regulate their production of the two glyoxalase enzymes themselves, but we know so far that zinc and insulin increase the production of glyoxalase-1 (29). This suggests that zinc, insulin, and glutathione are critical components of our defense against dicarbonyls and the AGEs they produce.
We should note here that since oxidative stress depletes glutathione, these findings suggest that promoting our antioxidant defense system is important to defending ourselves against the ravages of AGEs, and that since this whole process occurs within the cell, this constitutes further evidence that most of the action is happening intracellularly.
I'm sure everyone else is thinking this too at this point, but someone should say it: shouldn't F-3K and glutathione get married? F-3K is great at protecting proteins from “Too-Close” Glucose, and sure glucose might resent it and give himself a hernia huffing and puffing himself into a 3-deoxyglucosone, but when all is in working order this is of no consequence because glutathione and his glyoxalase defense squad simply wrestle this dangerous dicarbonyl down and rehabilitate him, restoring the peace. F-3K and glutathione are a perfect pair.
Does “High Blood Sugar” Cause the Formation of AGEs?
And thus, having gone halfway around the world and back we now return to the initial question I'd been asked, or at least the first half of it: does high blood sugar cause AGEs? I think we all agree that AGEs are increased in diabetes, but blaming all of the complications of diabetes on “high blood sugar” is like blaming all of the complications of familial hypercholesterolemia on “high cholesterol.”
Diabetes involves a lot more than high blood sugar. Most likely, increased concentrations of glucose and ketones, defective energy metabolism, defective insulin signaling, and oxidative stress lead to the increased production of and decreased detoxification of dicarbonyls. These dicarbonyls then form AGEs, defective degradation of AGE-modified proteins elevates their concentration further, and if the diabetes damages the kidneys, even the free AGEs released from degraded proteins will not be efficiently excreted. High blood sugar is a part of this, but only a part.
All this said, does the endogenous production of AGEs cause harm? In future posts, I will make a case that AGEs and their dicarbonyl precursors may emerge as key signaling molecules, but that in many situations they do indeed cause harm.
References
1. Henle T. Maillard Reaction of Proteins and Advanced Glycation End Products (AGEs) in Food. In Stadler RH, Lineback DR, eds. Process-Induced Food Toxicants: Occurrence, Formation, Mitigation, and Health Risks. Wiley Online Library, Published Online July 11, 2008.
2. Forster A, Kuhne Y, Henle T. Studies on absorption and elimination of dietary mallard reaction products. Ann NY Acad Sci. 2005;1043:474-81.
3. Hellwig M, Geissler S, Matthes R, Peto A, Silow C, Brandsch M, Henle T. Transport of free and peptide-bound glycated amino acids: synthesis, transepithelial flux at Caco-2 cell monolayers, and interaction with apical membrane transport proteins. Chembiochem. 2011;12(8):1270-9.
4. Hellwig M, Geissler S, Peto A, Knutter I, Brandsch M, Henle T. Transport of free and peptide-bound pyrraline at intestinal and renal epithelial cells. J Agric Food Chem. 2009;57(14):6474-80.
5. Henle T, Deppisch R, Beck W, Hergesell O, Hansch GM, Ritz E. Advanced glycated end-products (AGE) during haemodialysis treatment: discrepant results with different methodologies reflecting the heterogeneity of AGE compounds. Nephrol Dial Transplant. 1999;14(8):1968-75.
6. Bergmann R, Helling R, Heichert C, Scheunemann M, Mading P, Wittrisch H, Johannsen B, Henle T. Radio fluorination and positron emission tomography (PET) as a new approach to study the in vivo distribution and elimination of the advanced glycation endproducts N-carboxymethyllysine (CML) and N-carboxyethyllysine (CEL). Nahrung. 2001;45(3):182-8.
7. Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochm. J. 2003;375(Pt 3):581-92.
8. Rabbani N, Thornalley PJ. Quantitation of Markers of Protein Damage by Glycation, Oxidation, and Nitration in Peritoneal Dialysis. Perit Dial Int. 2009;29(Suppl 2):S51-6.
9. Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 200;275(50):39027-31.
10. Thornalley PJ, Rabbani N. Protein damage in diabetes and uremia — identifying hotspots of proteome damage where minimal modification is amplified to marked pathophysiological effect. Free Radic Res. 2011;45(1):89-100.
11. Agalou S, Ahmed N, Thornalley PJ, Dawnay A. Advanced glycation end product free adducts are cleared by dialysis. Ann NY Acad Sci. 2005;1043:734-9.
12. Thornalley PJ. Dicarbonyl intermediates in the mallard reaction. Ann NY Acad Sci. 2005;1043:111-7.
13. Ahmed N, Babaei-Jadidi R, Howeel SK, Beisswenger PJ, Thornalley PJ. degradation products of proteins damaged by glycation, oxidation and nitration in clinical type 1 diabetes. Diabetologia. 2005;48(8):1590-603.
14. Ahmed N, Thornalley PJ, Dawczynski J, Franke S, Strobel J, Stein G, Haik GM. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Invest Ophthalmol Vis Sci. 2003;44(12):5287-92.
15. Niyati-Shirkhodaee F, Shibamoto T. 1993. Gas chromatographic analysis of glyoxal and methylglyoxal formed from lipids and related compounds upon ultra-violet irradiation. J Agric Food Chem. 41;227-230.
16. Loil-Stahlhofen A, Spitelier G. [alpha]-Hydroxyaldehydes, products of lipid peroxidation. Biochim et Biophys Acta (BBA) — Lipids and Lipid Metabolism. 1994;1211(2):156-60.
17. Fu MX, Requena JR, Jenkin AJ, Lyons TJ, Baynes JW, Thorpe SR. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem. 1996;27(17):9982-6.
18. Van Schaftingen E, Delpierre G, Collard F, Fortpied J, Gemayel R, Wiame E, Veiga-da-Cunha M. Fructosamine 3-kinase and other enzymes involved in protein deglycation. Adv Enzyme Reglu. 2007;47:261-9.
19. Thornalley PJ. Glyoxalase I — structure, function, and a critical role in the enzymatic defense against glycation. Biochem Soc Trans. 2003;31(Pt 6):1343-8.
20. Bechara EJ, Dutra F, Cardoso VE, Sartori A, Olympio KP, Penatti CA, Adhikari A, Assuncao NA. The dual face of endogenous alpha-aminoketones: pro-oxidizing metabolic weapons. Comp Biochem Physiol C Toxicol Pharmacol. 2007;146(1-2):88-110.
21. Cai W, He JC, Zhu L, Chen X, Zheng F, Striker GE, Vlassara H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008;173(2):32736.
22. Casazza JP, Felver ME, Veech RL. The metabolism of acetone in the rat. J Biol Chem. 1984;259(1):231-6.
23. Casazza JP, Veech RL. The production of 1,2-propanediol in ethanol treated rats. Biochem Biophys REs Commun. 1985;129(2):426-30.
24. Casazza JP, Sohn DH, Park KS, Song BJ. Serum acetone and liver acetone monooxygenase activity in pregnant rats, fetuses, and neonates: reversible pre translational reduction of cytochrome P450IIE1 (P450IIE1) during pregnancy. Arch Biochem Biophys. 1994;309(1):111-6.
25. Beisswenger BG, Delucia EM, Lapoint N, Sanford RJ, Beisswenger PJ. Ketosis leads to increased methylglyoxal production on the Atkins diet. Ann NY Acad Sci. 2005;1043:201-10.
26. Nagai R, Nagai M, Shimasaki S, Baynes JW, Fujiwara Y. Citric acid inhibits development of cataracts, proteinuria and ketosis in streptozotocin (type 1) diabetic rats. Biochem Biophys Res Commun. 2010;393(1):118-22.
27. Abordo EA, Minhas HS, Thornalley PJ. Accumulation of alpha-oxoaldehydes during oxidative stress: a role in cytotoxicity. Biochem Pharmacol. 1999;58(4):641-8.
28. Dhar A, Dhar I, Desai KM, Wu L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes. 2011;60(30:899-908.
29. Ranganathan S, Ciaccio PJ, Walsh ES, Tew KD. Genomic sequence of human glyoxalase-I: analysis of promoter activity and its regulation. Gene. 1999;240(1):149-55.
You are the man, Chris!