
Is the “Receptor for AGEs (RAGE)” Really a Receptor for AGEs?
Is the “Receptor for AGEs (RAGE)” Really a Receptor for AGEs?

RAGE. It sounds pretty mad. Angry. And it's out to get you. How? By punishing you for eating overcooked food, igniting your tissues with the hellfire of inflammation and oxidative stress.
Or not. Maybe it's just there to make your brain grow. Welcome to the controversies of science department snack rooms and rarely published critical reviews.
In this blog post I'm going to present some evidence that the Receptor for AGEs (RAGE) is not a receptor for AGEs. And that RAGE's job isn't to punish you with a hellfire of inflammation and oxidative stress, regardless of whatever you did wrong, cooked food or no.
Advanced glycation endproducts (AGEs) are formed during chemical reactions between proteins and either sugars, lipid peroxidation products, or the breakdown products of sugars. By altering a protein's structure, they alter its function. It's possible that these alterations play a legitimate role in communication between cells and body systems (more on that in a later post), but it's also clear that at least in some cases, as in diabetes, the excessive glycation of mitochondrial proteins leads to enormous leakage of an oxidant called superoxide (1), which impairs blood vessel function (2).
According to conventional wisdom, AGEs cause their destruction not simply by altering the function of the proteins they modify, but also by binding to a specific receptor called “Receptor for AGEs,” abbreviated “RAGE.” These AGEs could be formed in the body or could be absorbed from food. Current reviews (3) claim that when AGEs bind to RAGE, this event precipitates a whole host of inflammatory cascades.
One of these critical cascades is the activation of the nuclear factor kappa-B (NFκB) pathway. NFκB will induce all kinds of inflammatory events in certain types of cells (although it is not inherently inflammatory), but it also stimulates production of RAGE. Thus, a vicious cycle supposedly ensues where RAGE stimulates NFκB and NFκB stimulates RAGE. Moreover, the oxidative stress supposedly invoked by RAGE causes more AGEs to be produced, and the incoming AGEs never cease binding RAGE and setting hellfire to our tissues.
The key word there is “supposedly.”
There are two problems with this theory. First, AGEs probably do not bind to and activate RAGE at all. Second, several studies suggest that activation of RAGE only stimulates direct inflammatory pathways when the agents used to activate it are contaminated with endotoxin, a known inflammatory component of certain bacterial cell walls!
“Supposedly” indeed!
Certain elements of the argument that AGEs do not bind to RAGE do indeed slip into the scientific literature. The most well known argument, which has been put forth even by PJ Thornalley (4), who has published a few hundred papers on glycation, is that test tube experiments only show AGE activating RAGE when the proteins are highly modified with 30-40 AGEs per protein. By contrast, proteins within the body only contain about one AGE modification per protein. Overcooked food may have highly AGE-modified proteins, but intestinal cells do not make much of the RAGE receptor and highly AGE-modified proteins are not absorbed intact.
You would think this argument would get more attention. After all, it seems implausible that an event would occur in the body when the components necessary for that event do not exist.
Some researchers nevertheless claim to have definitive evidence that AGEs bind to RAGE within live people and live animals, and these claims are generally not fully addressed. I took a look at the evidence myself, and let me say I was less than convinced.
The September, 2007 issue of Molecular Nutrition and Food Research published two opposing arguments addressing the question of whether AGEs really bind to and activate RAGE. In “Arguing for the motion: Yes, RAGE is a receptor for advanced glycation endproducts,” (5) Ramasamy, Yan, and Schmidt cited two studies (6, 7) claiming to provide definitive evidence that AGEs bind RAGE.
This is going to get a little technical, but please bear with me for a moment and I will explain this as best I can.
If your eyes glaze over, please skip to the part in red marked “the worst technical stuff is over” or the part in blue marked “the technical stuff is over.”
Here is the evidence from the first paper:

This is a picture of a blotting procedure where antibodies to RAGE were used to isolate RAGE and then antiobodies to a specific AGE called carboxymethyl-lysine (CML) were used to show that CML was bound to RAGE (in humans, column 1). Then, antibodies to CML were used to isolate proteins modified by CML and antibodies to RAGE were used to show that RAGE was bound to these proteins (in humans, column 3). The numbers on the left tell us how big these proteins are: the smaller the protein, the further towards the bottom its band (big black spot) is.
The first thing we can observe about this blot is that it is a mess. Some of these bands are so fat that you can't tell whether they are one band or five or six bands that are all merged into one. Usually when you publish a picture of one of these blots, you pick your best one. Scary.
The second thing is that, indeed, CML-modified proteins were bound to RAGE. This is shown in column 1 by the mere existance of one or more black bands. That is not very interesting, since virtually any protein can be modified by CML. All this suggests is that RAGE binds some or another protein, or is modified by CML itself.
The third thing is rather disconcerting. If we look at column 3, where each band represents what should be a RAGE receptor, we see there are at least six separate bands, one of which may actually be several bands all glommed together into one. There are only three variants of the RAGE receptor, so clearly this antibody was binding to all kinds of other stuff.
This makes you wonder, in how many other studies were the antibodies that were supposedly specific to RAGE actually binding all kinds of other things? This is especially troublesome when the antibodies are used to block the activation of RAGE and thereby show that an effect is due specifically to the activation of RAGE. The only conclusion one can come to is that all studies relying on antibodies to RAGE should be taken with a grain of salt unless they provide very rigorous evidence that their antibodies are highly specific to RAGE.
Here is the evidence from the second paper:
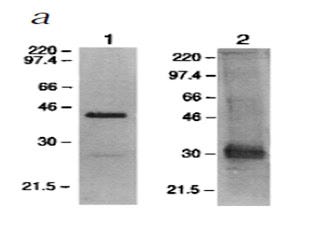
Here, the researchers purified RAGE from the blood of mice by using an anti-RAGE antibody. Then they labeled it with either an anti-RAGE antibody (left) or an anti-AGE antibody (right). Wow, “anti-AGE”? That's even less specific than an antibody to the specific AGE, CML. There are over a dozen types of AGEs and they can all occur on virtually any protein, so an “anti-AGE” antibody should be binding to lots and lots of things.
What is amazing about this blot is that, on the right, we see that this “anti-AGE” antibody seems to have been very specific for some singular, mysterious 30-kilodalton (kDa, a measure of mass or weight) “AGE.”
What could the identity of this mysterious 30-kDa “AGE” be?
A paper published three years earlier in 1995 (8) isolated RAGE from cow's lung. They provided very good evidence showing that they had isolated only one, single protein. Then they screened powdered lung tissue for RAGE-binding activity. Lo and behold, only two proteins bound to RAGE. And one of them was… *drumroll*… 30 kilodaltons! They sequenced the protein and showed it to be amphoterin, a protein that helps brain cells grow projections called neurites that they use to connect with one another, and which just happens to bind to RAGE fifteen times more effectively than even the highly modified AGEs that are only found in test tubes.
The other protein was 12 kDa. They didn't sequence it, but it was probably one of the small S100 proteins that are now known to bind to RAGE and weigh 10-12 kDa.
So, it seems that the evidence that “AGEs” bind to RAGE is pretty poor.
The Worst Technical Stuff is Over
The second problem is that activating RAGE only seems to directly stimulate inflammation when the activator is contaminated with endotoxin.
In 2004, Jessica Valencia and her colleagues at Novartis Institutes for Biomedical Research published two papers (9, 10) showing that when AGE-modified proteins were carefully prepared to avoid contamination with endotoxin, they did in fact bind to RAGE but they did not activate the NFκB pathway. By contrast, as expected, endotoxin itself did activate the NFκB pathway. When the AGE-modified proteins bound to RAGE, they did cause small changes in the expression of nine genes, but none of them were inflammatory and the only pattern that emerged were that a few of them were related to the ability of cells to “go forth and multiply.”
More recently, researchers showed the same thing with amphoterin, which is now called high mobility group 1b protein (HMGB1) (11). They purchased two lots of HMGB1 from Sigma and one from R&D systems. One of the lots from Sigma was contaminated with high levels of endotoxin and the other two lots were not. Here are their results:
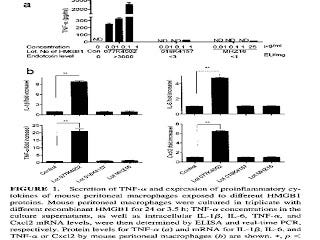
The top graph shows that the contaminated lot, on the left, produced a dose-dependent increase in the expression of tumor necrosis factor alpha (TNF-alpha), while the other two lots did not. The bottom four graphs showed that, with a single dose, only the contaminated lot (second bar in each graph) increased the expression of TNF-alpha and three other inflammatory molecules. For a clearer view of this graph, you can check out the original here.
The Technical Stuff is Over
So, it appears that RAGE is not out to get us after all.
What, then, is it there for?
Here's my take, based on the evidence as reviewed in reference 3. The main activators of RAGE within living organisms are HMGB1 and the small S100 proteins. When HMGB1 binds to RAGE it promotes cell proliferation and migration, plays a role in embryonic development and tissue repair, and plays an essential role in the development and function of the innate immune system. When S100 proteins bind to RAGE, they stimulate cell survival. It is possible that at very high concentrations in certain pathological conditions they may promote inflammation, but if this is the case it seems to be the exception rather than the rule.
Thus, if we delete the RAGE gene in mice, they benefit from reduced migration of inflammatory cells into the blood vessels that will oxidize LDL when studied in a model of atherosclerosis, reduced enlargement of the kidney when studied in diabetic models, and reduced inflammation in models involving overstimulation of the immune system such as colitis or septic shock. On the other hand, HMGB1 binding to RAGE enhances the regeneration of the heart after a heart attack, and the regeneration of nerves after injury, showing a general role in tissue repair.
The above review concluded that “although mutations deleting RAGE function may not be compatible with life and therefore not found in cohort studies, the fact that [mice with the RAGE gene deleted] are viable and do not show an overt phenotype [either] contradict[s] this hypothesis or point[s] to important differences between mice (kept in a sterile environment) and human diabetes.”
I propose that the need to keep these mice in a sterile, or at least specific pathogen-free, environment, provides an obvious explanation for why these “beneficial” deletions of the RAGE gene are not compatible with life outside of the laboratory, where exposure to pathogens is constant.
Thus, RAGE is not out to get us. It's out to turn us into symmetrical human beings with a working brain and immune system, and to help us repair our tissues when we are injured.
Can it be involved in pathological processes? Sure. So can the rest of the human body. But the human body is a good thing.