
How Conflating the Lipid Hypothesis With the Diet-Heart Hypothesis Led to the Public Condemnation of Bacon, Butter, and Eggs
How Conflating the Lipid Hypothesis With the Diet-Heart Hypothesis Led to the Public Condemnation of Bacon, Butter, and Eggs

In my last two posts, I argued for the importance of distinguishing the lipid hypothesis, which holds that high concentrations of cholesterol in the blood cause heart disease, from the diet-heart hypothesis, which holds that high amounts of saturated fat in the diet cause heart disease.
If you missed them, you can read them here:
Ironically, Gary Taubes and Daniel Steinberg — arguing for nearly opposite views of the history of these controversies — both make this exact point in their respective books.
There are a few things, however, that Taubes doesn't tell us that make this distinction even more important than we would think it is after reading his book. These will be discussed below.
In Good Calories, Bad Calories, Taubes makes a critical point about the public reception of the 1984 Coronary Primary Prevention Trial (CPPT) that I have made in numerous places, including my interview with Jimmy Moore and my most recest Wise Traditions lecture.
The CPPT tested the effect of a drug that lowers levels of cholesterol in the blood, cholestyramine, on the risk of heart disease. But what conclusion did the media push on the American people?

Image hosted at Time Magazine
{kind=link}
In an article running the headline “Hold the Eggs and Butter,” Time Magazine announced that “cholesterol is proved deadly, and our diet may never be the same.”
The trial had been designed “to test the ‘lipid hypothesis,' i.e., the hypothesis that intervention to reduce serum cholesterol levels does reduce risk of clinically manifest coronary artery disease,” in the words of Daniel Steinberg, who chaired the planning committee. But Time was making a conclusion about the diet-heart hypothesis.
Taubes doesn't use these exact terms, but he makes the same point (p. 57-8; his italics, my bold):
Pete Ahrens called this extrapolation from a drug study to a diet “unwarranted, unscientific and wishful thinking.” Thomas Chambers, an expert on clinical trials who would later become president of the Mt. Sinai School of Medicine in New York, described it to Science as an “unconscionable exaggeration of all the data.” In fact, the LRC investigators acknowledged in their JAMA article that their attempt to ascertain a benefit from diet alone had failed.
Rifkind later explained the exaggerated claims. For twenty years, he said, those who believed in Keys's hypothesis had argued that lowering cholesterol would prevent heart attacks. They had spent hundreds of millions of dollars trying to prove it, in the face of extreme skepticism. Now they had demonstrated that lowering cholesterol had reduced heart-disease risk and maybe even saved lives. They could never prove that cholesterol-lowering diets would do the same — that would be too expensive, and MRFIT, which might have implied such a conclusion, had failed — but now they had established a fundamental link in the causal chain from lower cholesterol to cardiovascular health. With that, they could take the leap of faith from cholesterol-lowering drugs to cholesterol-lowering diets. “It's an imperfect world,” Rifkind said. “The data that would be definitive is ungettable, so you do your best with what is available.”
With publication of the LRC results, the National Heart, Lung, and Blood Institute launched what Robert Levy called “a massive health campaign” to convince the public of the benefits of lower cholesterol, whether by diet or drug, and the media went along. Time reported the LRC findings in a story headlined “Sorry, It's True. Cholesterol Really Is a Killer.” The article about a drug trial began, “No whole milk. No butter. No fatty meats. Fewer eggs . . .”
Daniel Steinberg, the chief architect of the trial, made the same point in his book, The Cholesterol Wars: The Skeptics vs. the Preponderance of the Evidence (p. 1, 37; his italics, my bold):
The “lipid hypothesis” . . . relates to blood lipids, not dietary lipids, as the putative directly causative factor. Although diet, especially dietary lipid, is an important determinant of blood lipid levels, many other factors play important roles. Moreover, there is a great deal of variability in the response of individuals to dietary manipulations. Thus, it is essential to distinguish between the indirect “diet-heart” connection and the direct “blood lipid-heart” connection. Failure to make this distinction has been a frequent source of confusion. . . .
In the 1950s and 1960s, before effective cholesterol-lowering drugs became available, the only way to lower blood cholesterol was to manipulate the diet. Hence, there emerged a tendency to short-circuit the “diet-blood cholesterol-heart disease” problem by omitting the “blood cholesterol” and talking about the “diet-heart” problem. As a result, many observational studies examined the correlation between dietary patterns and the incidence of coronary heart disease without asking whether the dietary patterns considered actually caused a significant change in blood cholesterol levels. This resulted in a great deal of confusion and misunderstanding.
Taubes thus makes the distinction to point out the invalidity of extrapolating from a drug trial to a dietary recommendation, while Steinberg makes the distinction to argue that the lipid hypothesis, despite the strength of the evidence behind it, took so long to be accepted because of the confusion generated by conflating it with the diet-heart hypothesis, whose evidence was much weaker.
It is somewhat ironic, then, that the authors of the CPPT and the panelists of the NIH Consensus Conference that shortly followed directly facilitated this confusion by making the extrapolation from drugs to diet themselves. After all, Steinberg himself chaired the planning committee of the trial and chaired the subsequent conference.
In the JAMA paper in which they initially reported their results, the authors made the remarkable claim that the results could not be extrapolated from cholestyramine to other drugs because other drugs may have a different mechanism of action, but that given the inability of providing solid evidence for dietary recommendations, the results should be considered supportive of dietary changes that would lower cholesterol levels.
As Taubes chronicles, Basil Rifkind led the way in making this extrapolation in the popular press and even told Time that the extrapolation from drugs to diet was “indisputable.” Rifkind was an even higher authority over the CPPT and the subsequent Consensus Conference than Steinberg. He took over for Robert Levy partway through the trial as the head of the NIH's Central Program Office and was essentially the lead director of the trial. Steinberg writes kindly of his necessary leadership skills but notes the frustration many in the trial had with the “no-discussion-allowed” approach he often had to take to get the job done .
Rifkind was the Chief of the Lipid Metabolism-Atherogenesis Branch of the National Heart, Lung and Blood Institute, which proposed the NIH Consensus Conference, and was the man who invited Steinberg to chair the conference. The appointment had to be approved by an independent branch of the NIH, the Office of Medical Applications of Research, but Rifkind was clearly a key player.
While Steinberg may have been the first to use the term “lipid hypothesis” in print, Edward H. “Pete” Ahrens Jr. popularized it soon after and was quickly cited as having originally defined it.
Pete Ahrens had a much greater sense of the complexity involved in supporting or falsifying a biological hypothesis than most other supporters or skeptics of the lipid hypothesis, and had applied a much greater level of scientific caution in this arena. When he originally defined the lipid hypothesis, he wrote about the need to differentiate between diet and drug trials, between people whose high cholesterol was determined by genetics or environment, and between important differences between populations, genders, and individuals. His presentation at the 1984 Consensus Conference was entitled “The lack of appropriateness (at this time) of public health measures to change American dietary habits.”
In fact, Ahrens gave this presentation immediately following another entitled “The appropriateness of public health measures to change American dietary habits to reduce blood cholesterol.” Perhaps the NIH used the word “consensus” to describe this conference for the sake of irony.
Nevertheless, in reading Good Calories, Bad Calories it is easy to come away with the sense that distinguishing between the lipid hypothesis and the diet-heart hypothesis is mostly useful for briefly demonstrating the stupidity of the medical and scientific establishments and is otherwise an unimportant matter of semantics that bears little relevance since there is such little evidence to support either hypothesis. After all, Taubes spends a page or two arguing against extrapolating from a drug trial to a dietary recommendation, but ultimately lumps all of this together as “The Fat-Cholesterol Hypothesis.”
There are a number of things, however, that Taubes leaves out of the story that make this distinction all the more important.
Consider for a moment his description of the actual results of the trial (p. 57):
In January 1984, the results of the trial were published in The Journal of the American Medical Association. Cholesterol levels dropped by an average of 4 percent in the control group — those men taking a placebo. The levels dropped by 13 percent in the men taking cholestyramine. In the control group, 158 men suffered nonfatal heart attacks during the study and 38 men died from heart attacks. In the treatment group, 130 men suffered nonfatal heart attacks and only 30 died from them. All in all, 71 men had died in the control group and 68 in the treatment group. In other words, cholestyramine had improved by less than .2 percent the chance that any one of the men who took it would live through the next decade. To call these results “conclusive,” as the University of Chicago biostatistician Paul Meier remakred, would constitute “a substantial misuse of the term.”
Taubes places so little emphasis on the findings related to heart disease that he doesn't even calculate the relative decrease in risk for us. Instead, he jumps immediately from the raw heart disease data to the absolute risk of dying from any cause. The trial was not designed to answer a question about total mortality and indeed it would have been statistically underpowered to do so.
Taubes also ignores the basic design of the trial. Before randomizing the participants to receive either the drug or the placebo, the investigators first stratified them into eight different levels of risk based on their LDL-cholesterol, electrocardiogram results, age, blood pressure, and tobacco use. Taubes reports his own statistics calculated from the raw data without making any adjustments for which strata the people who died belonged to, not even adjusting for age, which obviously makes people more likely to die. With the appropriate adjustments, the reduction in total mortality was still not statistically significant, but it was 7 percent, 35 times greater than what Taubes reports.
Perhaps one could mount an argument against their method of stratification, but it would be more appropriate to make that argument than to write as if the stratification didn't exist.
When addressing a scientific test of a cause-and-effect phenomenon, the central question is whether the relative risk of the effect changes when we manipulate the putative cause. In this case, we want to manipulate the level of cholesterol in the blood and see if it changes the risk of heart disease.
The absolute risk becomes more relevant when we are trying to decide whether to prescribe the drug as a physician, or whether to take it as a patient. In that case we want to know the number needed to treat, which tells us as a physician how many people we need to give the drug to in order to save a life, or as a patient, what the likelihood is that we will personally benefit from taking the drug. In these cases, we would also want to know more about total mortality, because we do not want to take or prescribe drugs that will cause as many problems as they resolve.
But to identify a specific cause-and-effect pathway, we want to know if the treatment manipulates the relative risk of a specific disease.
As a scientific question of whether reducing blood cholesterol levels can reduce the relative risk of heart disease, the Coronary Primary Prevention Trial proved remarkably supportive of the lipid hypothesis.
The drug decreased the risk of the primary endpoint, definite nonfatal and fatal heart attacks, by 19 percent. Analyzed separately, the treatment decreased the risk of nonfatal heart attacks by 19 percent and fatal heart attacks by 24 percent. The treatment also reduced the risk of a new positive exercise test by 25 percent, angina by 20 percent, and coronary bypass surgery by 21 percent.
These results were statistically significant at a one-tailed p-value of less than 0.05, which means that we can be 95 percent confident that the treatment was effective and that the difference was not due to chance. The significance of the term “one-tailed” means that if we had been equally concerned with the question of whether the drug may have had the opposite effect, we could have only been 90 percent confident that the difference was genuine.
The investigators, of course, were initially hoping to get a more rigorous demonstration of statistical significance, but when we appreciate exactly what a horror it was to take both the drug and the placebo, we can easily see why the results came out less impressive. And, in fact, this difficulty actually allows us to see quite powerful evidence of a dose-response that supports the lipid hypothesis even further.
Of course, eventually I will discuss why these results do not support the “infiltrative” lipid hypothesis when considered in the light of the total evidence.
Taubes refers to the placebo as a “placebo pill” and gives the reader the impression that this intervention was rather straightforward. In point of fact, however, neither the drug nor the placebo came in the form of a “pill.” They both came in the form of six packets of sandy powder that had to be taken two packets at a time, three times a day, by stirring into juice or water. The placebo consisted of polymer beads the same size and color but without any drug activity. Both the treatment and the placebo had high rates of gastrointestinal side effects, though most were mild and could be managed easily with other medications.
This trial had to get almost 4,000 men to take this stuff three times a day every day for 7-10 years!
As a result, many men readily admitted that they stopped taking the drug altogether within a few weeks of starting the trial. Others simply reduced the amount of drug they took. During the first year, the men took an average of 4.2 packets a day instead of the six packets that the protocol called for. By the end of the study, the men were taking on average less than four packets per day.
The investigators reported the results for everyone, regardless of how compliant they were. Today we would call this an intention to treat analysis, which allows policy makers to determine how effective a treatment will be knowing that many people will not comply with it. Often in trials nowadays authors will present these results side-by-side with a per protocol analysis, which shows the results in the fully compliant subjects to estimate what the actual effect of the drug is.
Thus, they intended the treatment to lower cholesterol levels by 24 percent, but it only lowered cholesterol levels by 9 percent, simply because many people in the treatment group were taking lower doses of the drug than the protocol intended.
In people who took five or more packets per day, however, total cholesterol decreased by 25 percent and LDL-cholesterol decreased by 35 percent.
This variation in compliance allowed the investigators to look for a dose-response between compliance, reductions in total and LDL-cholesterol, and reductions in heart disease risk. Indeed, they found one, and published it in a second JAMA article. It was statistically significant at p<0.001, meaning that we can be 99.9 percent confident the results are genuine and not due to chance.
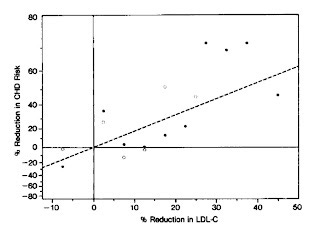
In this graph, the authors divided the participants into blocks representing successive five-percent reductions in LDL-cholesterol. The black circles are men in the treatment group while the white circles are men in the placebo group. As we move to the right, the men experienced greater reductions in LDL-cholesterol. As we move upward, the men experienced greater reductions in heart disease risk. There is quite clearly a dose-response.
The stray dot at the extreme right may perhaps indicate that there is such a thing as “too low,” but we can clearly see that those who took enough cholestyramine to lower LDL-cholesterol more than 25 percent had much greater reductions in heart disease risk than those who took less.
Of course, this data would be more impressive if the investigators had actually randomized the men to receive different doses. After all, people who are more compliant are often more health-conscious. Nevertheless, adherence was only associated with risk reduction in the treatment group, not in the placebo group. Likewise, change in LDL-cholesterol was only associated with risk reduction in the treatment group. This very strongly suggests that the greater reduction in risk was a result of the greater dose of the drug.
Moreover, when the number of packets of the drug the men took was considered in a statistical model with the degree of cholesterol-lowering achieved, the packet count became statistically insignificant (p>0.85), while the significance of the cholesterol-lowering effect persisted (p<0.001). This supports even more strongly that this dose-response was a genuine effect of the drug.
Supporting or falsifying a biological hypothesis, however, is incredibly complex. Cholesterol-lowering drug trials are all confounded by the fact that all of the pharmacological agents used in them lower cholesterol levels indirectly and most of them do so without complete specifcity for that effect. Cholestyramine does so more specifically than statins do, but no more directly.
A few months after the Coronary Primary Prevention Trial was published, Steinberg's group published a paper in the Proceedings of the National Academy of the Sciences that showed that the oxidative destruction of the LDL particle was dependent on oxidation of polyunsaturated fatty acids that proponents of the diet-heart hypothesis were telling us to eat. Steinberg's group had already provided data strongly suggesting that the concentration of cholesterol in the blood has little or nothing to do with the risk of heart disease, while oxidative degeneration of LDL particles has an awful lot to do with it.
Indeed, there is a wealth of evidence giving the LDL particle a central role in the development of atherosclerosis. None of this evidence, however, specifically indicts the concentration of cholesterol associated with that particle. The totality of the evidence paints a very different picture. I will review some of this powerful evidence in the next blog post.