
What Can Familial Hypercholesterolemia Mortality In the 19th Century Teach Us About Genetics?
What Can Familial Hypercholesterolemia Mortality In the 19th Century Teach Us About Genetics?

Not much, simply because the data are so poor. However, let's take a look at some data for the very weakly supported hypothesis that people in the 19th century with familial hypercholesterolemia lived longer than other people and perform a little “thought experiment” to see what it could teach us about genetics if it were actually true.
I was going to include this in my “New Genetics” series but it turns out that the data just aren't convincing enough to give this post that kind of status. Let's take a look.
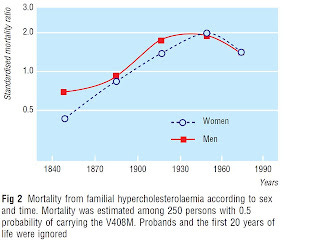
Here's a chart from a paper by Sijbrands et al. showing estimated mortality in people with heterozygous familial hypercholesterolemia, compared to mortality in the Dutch population in which they lived. These are people who have one copy of a defective gene for the LDL receptor, which brings LDL particles into cells. They also have one normal copy of the gene.
The line marked “1.0” indicates mortality equal to the rest of the population. Where the line sinks below this point, mortality was lower. Where it rises above this point, mortality was higher.
In his book The Cholesterol Myths, Uffe Ravnskov (MD, PhD) cites this study and claims that “those living in the 19th century had a lower mortality than the general population. After 1915 the mortality rose to a maximum between 1935 and 1964, but even at the peak, mortality was less than twice as high as the general population.”
Umm, almost. Except the authors don't say it quite like this. A sidebar in the paper summarizes that “standardised mortality ratio was normal in the 19th century and rose to a peak in the 1930s to 1960s,” and the authors venture out on a limb to suggest that “in the 19th century, mortality seemed lower than in the general population.”
And for good reason.
First, this is mortality for people with the gene that is estimated from the mortality data among first-degree relatives of people known to have transmitted the gene to living descendants. These people thus only had a fifty percent chance of having the gene. Among people actually known to have the defective gene, mortality was only lower during a single time period, 1830-1869, and this was based on a single person's death.
Second, there was never any time period during which there was a statistically significant decrease in mortality. The mortality difference became statistically significant after 1935 and by that point was increased.
Why might that be? Well, if in fact mortality was lower during the nineteenth century, this may be because rates of infectious disease were high, and there is some evidence that high cholesterol levels protect against systemic infections.
There might be another reason, however. Let's take a look at their pedigree.
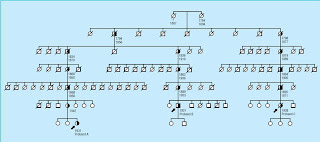
They had found three distantly related people with a common ancestral couple who died in the early 1800s, and then they traced that couple's descendants.
What else increased over the centuries besides estimated mortality? How about their sample size?
Let's take a look at the number of deaths their data is based on, and we will see the same trend.

You'll be able to see the data a little better if you click on the table. As the two centuries progressed, the mortality rates for people actually known to have familial hypercholesterolemia were based on a single death in the first few decades, then three deaths, then four, then eleven, and then finally thirty. Among the people with a fifty percent chance of having a copy of the defective gene, the mortality rates were based on three deaths, then seven, then eleven, then twenty-three, then twenty-six, and then finally seventy.
The possibility that random variation and poor sampling was a major factor for the nineteenth century can be seen for the period between 1870 and 1904, where a 13 percent decrease in risk is observed among those with a fifty percent chance of having the gene, suggesting the gene might actually decrease the risk of dying about 26 percent, while the mortality estimate based off people known to be affected was increased by 20 percent. These putative “effects” are diametrically opposed to one another, and neither of them are statistically significant.
So, it could be that mortality was lower in the 1800s and higher in the 1900s. Or, it could be that sample size increased over these two centuries. Consequently, the accuracy of the estimate increased, until finally after 1935 a statistically significant doubling of mortality was observed.
(Of course this is total mortality, and heart disease mortality estimates would be much higher.)
In any case, if we accept the poorly supported hypothesis that mortality was lower in the 19th century among those with heterozygous familial hypercholesterolemia for the purpose of this thought experiment, what can it teach us about genetics?
What it teaches us is that when the environment is homogeneous, genetic studies will always overestimate the effect of genetics and underestimate the effect of the environment.
As I pointed out in my post, “Lack of Correlation Does Not Show Lack of Causation,” one reason we might not observe a relationship with something even if it is a true and active cause of the effect we are looking at is simply because there is not enough variation. Ned Kock has a great post about this with another little experiment showing how a study with too little variation in smoking habits could lead to the false conclusion that lung cancer is “genetic.”
If we were to follow the absurd but entirely normal and ubiquitous habit of calling a variant in the DNA sequence of a gene (precisely termed an allele) that predisposes for a particular effect “a gene for _____,” we would call the gene associated with familial hypercholesterolemia the gene for death.
That should be our first lesson — just how stupid this practice of naming genes actually is. The gene actually codes for the LDL receptor. That's its physiological function. But if we follow the colloquial terminology that talks about “the gene for cancer,” “the gene for diabetes,” or “the gene for anger,” or heck, even the professional and scientific terminology that calls a gene an “oncogene” instead of a “cell cycle gene,” then we would call this gene the gene for death, because it influences mortality.
Note that it would be entirely sensible to refer to it as a gene for familial hypercholesterolemia, even if it would be much more accurate to refer to it as an allele for familial hypercholesterolemia, because this disease is monogenic. That is, it is caused very specifically by this single defective gene.
Most of the time when we say “the gene for _____,” we are referring to traits that are both polygenic and affected by the environment. Thus, the practice is analagous to calling a defective LDL receptor gene a gene for death because the risk of death is both polygenic and affected by the environment. It would also be analagous to calling it “a gene for high cholesterol,” but the absurdity in this case is less obvious so I'm using the phrase a gene for death for the sake of its absurdity to make my point more obvious.
The second thing we see is that its effect can be dependent on the environment, but if the environment doesn't vary enough, the gene seems to be the sole determinant.
Let's say that an environment favored infectious disease in the 1800s but favored heart disease in the 1900s. Perhaps high cholesterol protects against infection but oxidative destruction of the LDL particle promotes heart disease. Low LDL receptor activity will promote both high blood cholesterol and oxidative destruction of the LDL particle, so it could have had the opposite effect in the 1800s as it had in the 1900s. A study conducted in the 1900s among living people would have had absolutely zero ability to detect the effect of the environment.
The lesson? Genetic studies will tend to overestimate the effect of genetics because of homogeneity in the environment. If the allele is associated with diabetes, it might be dietary homogeneity — too much vegetable oil and refined carbs wherever you look. If the allele is associated with personality, it could be cultural homogeneity — a dysfunctional public school system, or homogeneous religious or cultural beliefs about or attitudes towards some particular thing.
As for familial hypercholesterolemia and its varying effect on mortality over time, it's an interesting hypothesis, but the data are too sparse to support or refute it.
Read more about Chris Masterjohn, PhD, here.