
Genes, LDL-Cholesterol Levels, and the Central Role of LDL Receptor Activity In Heart Disease
Genes, LDL-Cholesterol Levels, and the Central Role of LDL Receptor Activity In Heart Disease

Are high concentrations of LDL-cholesterol a major cause of heart disease? If we are a proponent of the “lipid hypothesis,” we say yes. If we are a “cholesterol skeptic,” we say no — total cholesterol, LDL-cholesterol, LDL particles, triglycerides, and other blood lipids have little or nothing to do with heart disease.
I believe both of these positions are wrong.
I think the evidence for the central role of the of the LDL particle in the development of atherosclerosis is overwhelming. However, I believe the evidence is very strong that the LDL particle is a victim rather than a perpetrator in the process, and that it is the oxidative destruction of the particle itself rather than the concentration of cholesterol within it that is the culprit. In this post, we will explore some important pieces of this evidence, and consider whether measuring blood cholesterol is meaningful. In the next post in this series, I will consider the possibility that leptin signaling and thyroid hormone are central governors of the risk of heart disease.
Let's consider for a moment this graph from the Framingham Study (1).
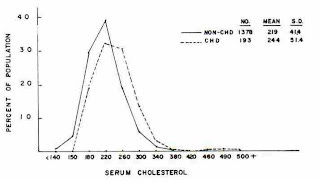
The dotted line represents the people who developed coronary heart disease, while the solid line represents those who remained heart disease-free. There are several legitimate points we could make about this graph on either side of the “lipid hypothesis” argument.
If we are a “cholesterol skeptic,” we might point out that the dotted line is only shifted to the right marginally, meaning that people who developed heart disease only had slightly higher cholesterol, on average, than people who did not. We might also point out that the vast area in the middle is shared by both those with and without heart disease. In fact, the solid line represents about seven times as many people as the dotted line. That means that the vast majority of people who developed heart disease developed it with cholesterol levels similar to those of seven times as many people who remained free of heart disease.
But let's look a little more closely at the graph. What else can we learn from it? Over to the right, we see a hump in the dotted line where everyone has heart disease. On the left, we see an area below 150 mg/dL where no one has heart disease.
Who are these people? And can they teach us anything about the cause of heart disease?The Genetic Evidence — LDL Receptor Activity
Many of the people in the hump on the right are undoubtedly people with familial hypercholesterolemia. Familial hypercholesterolemia results from a genetic defect in the LDL receptor, which is the receptor required to bring the LDL particle into cells. It can come in two forms. People who inherited only one defective copy of the gene from either their father or mother are heterozygous for the disease, while people who inherited a defective copy from both parents are homozygous for the disease.
People who are homozygous for familial hypercholesterolemia can develop atherosclerosis and coronary heart attacks in infancy (2–3).

In the third edition of The Metabolic Basis of Inherited Disease (1972), Fredrickson and Levy reported a South African child with the homozygous form of this disease who died of a heart attack when only 18 months old. The oldest survivor in their case series died at age 27 after his third heart attack.
Thus, while most people become at risk for heart disease after the age of 60, people with two defective copies of the LDL receptor gene have this risk shifted back to the ages of 0-30 and are pretty much guaranteed to die from atherosclerosis or related complications early in life.
People who have only one copy of the defective gene have the risk of heart disease shifted instead to the 30-60 age range, and their age- and sex-adjusted risk of heart disease is dramatically increased (4).
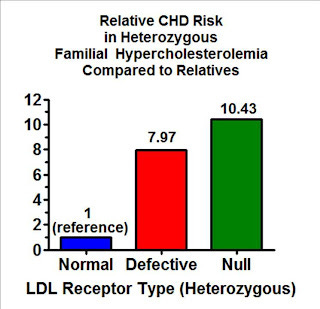
If we amass groups of people who have one copy of any number of different mutations that result in a defective LDL receptor and compare them as a group to their unaffected relatives, adjusting for age and sex, those with a mutation that decreases the effectiveness of the receptor have a four-fold elevated risk of heart disease, while those who have a mutation that eliminates the gene's ability to code for an LDL receptor entirely have a six-fold elevated risk.
But if we specifically match each person with a defect to their own relatives so that we control for any other genetic and environmental factors they share in common, those with a copy of a partially defective gene have an eight-fold increase in risk and those with a completely defective gene have a more than ten-fold increase in risk.
Now, who might those people be on the other side of the Framingham graph who have low cholesterol levels and zero incidence of heart disease? Perhaps some of them have a genetic defect in the PCSK9 gene, which codes for the enzyme that degrades the LDL receptor. These people have a genetic defect that increases LDL receptor activity instead of decreasing it. This is essentially the opposite of familial hypercholesterolemia.
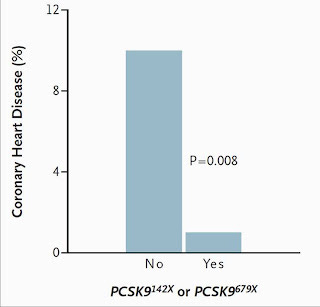
Those who have a “nonsense” mutation that deletes the enzyme have a whopping 88 percent reduction in the risk of heart disease (5)! That's virtual abolition!
Thus if we look at the genetic evidence, we see a dose-response effect of LDL receptor activity. Mutations that increase its effectiveness virtually abolish the risk of heart disease. One copy of a mutation that decreases its effectiveness pushes the risk back from the over-60 age range to the 30-60 age range and raises the risk eight- to ten-fold. Having two copies of a defective gene pushes the risk back to the 0-30 age range, virtually guarantees an early death, and can produce heart attacks in infancy.
Can these genetic mutations tell us anything about what causes heart disease in the rest of us?
I would argue that this is incredibly powerful evidence that we can and should use to learn about the cause of heart disease in people without these mutations, so long as other forms of evidence from mutation-free populations are consistent with and supportive of the hypothesis we develop.
Why? Mutations in single genes that affect single traits are the closest form of evidence we have to a lifelong experiment.
Genetic mutations, however they develop, are randomly sorted and distributed to the sex cells that eventually form new human beings. Thus, this type of evidence, although observational, is much closer to a “natural” randomized experiment than a typical observational study, and it's the only form of such evidence that can be carried out over a lifetime.
Sometimes genetic variants can be linked to variants in other genes nearby on the chromosome. However, this cannot possibly account for the genetic evidence presented here thus far, because the study referenced above included people with nine independent mutations in the LDL receptor, four of which were present in substantial numbers, and the PCSK9 gene isn't even on the same chromosome!Furthermore, the fact that adjusting for family ties increased rather than decreased the risk estimate for heart disease among people with familial hypercholesterolemia in the above study provides further confirmation that the increase in risk is due specifically to the LDL receptor mutations.
Thus, the genetic evidence suggests that LDL receptor activity plays a central role in governing the risk of heart disease.
While it would be wrong to consider this genetic evidence separately from other forms of evidence without trying to synthesize the totality of the evidence into a coherent theory, it would likewise be wrong to assume that the findings of these genetic studies transfer to humans without these mutations. It is possible that the range of LDL receptor activity in “normal” people just isn't broad enough to impact heart disease risk, or even that there is some type of funny-shaped risk curve that looks like this:
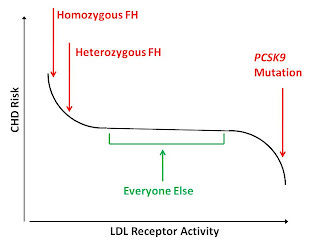
Can we find evidence supporting a role for LDL receptor activity among those who do not have these mutations? If so, this would allow us to synthesize these different forms of evidence into a coherent theory about the importance of LDL receptor activity.
Modest Evidence From Drug Trials Suggests That Increasing LDL Receptor Activity Lowers CHD Risk
As it turns out, we already have evidence from randomized, controlled drug trials showing that drugs that increase LDL receptor activity decrease the risk of heart disease. Both cholestyramine and statins lower blood levels of LDL-cholesterol by increasing LDL receptor activity (6).
Here are two slides from my most recent Wise Traditions lecture, “Heart Disease and Molecular Degeneration: The New Paradigm,” showing how this occurs. You can click on them to enlarge them if necessary.
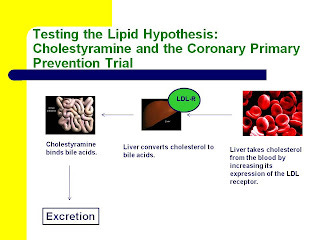
Cholestyramine binds bile acids and causes their excretion. As a result, the liver makes more bile acids from cholesterol. The level of free cholesterol in the liver thus declines, and the liver increases its expression of the LDL receptor in order to take in more cholesterol from the blood.

Statins target cholesterol synthesis in the liver. By inhibiting cholesterol synthesis, they decrease the level of free cholesterol in the liver just like cholestryamine. Consequently, the liver increases its expression of the LDL receptor in order to take in more cholesterol from the blood.
As I pointed out recently in my post, “Conflating the Lipid Hypothesis With the Diet-Heart Hypothesis Led to the Public Condemnation of Bacon, Butter, and Eggs,” the Coronary Primary Prevention Trial (CPPT) provided very convincing evidence that cholestyramine reduces the risk of heart disease, with strong, though perhaps not definitive, evidence for a dose-response.
Virtually everyone agrees, cholesterol skeptic or lipid hypothesis proponent, that statins reduce the risk of heart disease and related mortality at least in certain specific well-studied populations, such as middle-aged white males with established heart disease, but the evidence that would be relevant here, and comparable to the Coronary Primary Prevention Trial with cholestyramine, is whether they prevent heart disease in people who are initially heart disease-free.
A recent Cochrane Review pooled the results of nine statin trials conducted in people without established heart disease and found a 28 percent reduction in the relative risk of cardiac events (7).
The authors also warned, however, that most of the trials did not report adverse effects at all, that in some trials the investigators may have exaggerated their effects by stopping the trial short when the results looked good, that Big Pharma sponsored all of the trials except one, that the populations tended to be white, male, and middle-aged, that investigators have downplayed potential side effects such as diabetes and avoided even looking for others such as cognitive impairment, that the reporting of details was so poor that the data were ultimately “impossible to disentangle,” and that if they had loosened their criteria of which trials to include their analysis could have shown no benefit at all.
Thus the evidence from statins is somewhat supportive, but should probably be taken with a grain of salt.
Unfortunately, it is currently impossible to provide a single, definitive test of whether increasing LDL receptor activity in members of the general population can reduce their risk of heart disease. Not only are statin trials usually too short (one to five years, compared to 7-10 year followup in the CPPT), but all of these drugs increase LDL receptor activity indirectly and without complete specificity.
Statins inhibit the synthesis of mevalonate, a compound that eventually, far down in the biochemical pathway, gets converted to cholesterol. But our cells use mevalonate for many other things, and thus statins have many “pleiotropic” effects. One such effect is to decrease the activation of a little enzyme called Rho, an enzyme that is part of the stress response and almost certainly contributes to both atherosclerosis and thrombosis. On the other hand, statins also decrease the synthesis of coenzyme Q10, a compound that likely protects against heart disease. Thus, statins are likely to have opposing effects on the risk of heart disease through different mechanisms.
All of these effects result from the inhibition of mevalonate synthesis, meaning they should all correlate with one another. Thus, the question of whether the effect of statins on mortality correlates with their effect on cholesterol levels is meaningless.
Cholestyramine's effect on LDL receptor activity is more specific than the effect of statins, and the results of the CPPT are more convincing than those of the statin trials, but cholestyramine's effect is still indirect. There is no way to definitively separate its effect on the LDL receptor from its effect on many other parameters, such as the amount of cholesterol degraded and excreted from the body, or potential effects on the absorption of other compounds from the gut.As I will discuss in a future post, thyroid hormone is the other central governor of LDL receptor activity besides the cholesterol level within the cell. While the cell regulates its expression of the LDL receptor for the sake of maintaining its own constant state of free cholesterol, thyroid hormone communicates to the cell that the organism is in a state of abundance and can dispose of that cholesterol by making bile acids and reproductive hormones, which enhance digestion, strength, and fertility.
Yet can we conduct a specific test of the hypothesis that LDL receptor activity is a central governor of heart disease risk by conducting a randomized, controlled trial using thyroid hormone or using natural means of normalizing thyroid hormone status? Of course not. Thyroid hormone has all kinds of metabolic effects and is far less specific to LDL receptor activity than cholestyramine, and even less specific than statins. Of course, normalizing thyroid hormone status would improve digestion, cognitive impairment, and other parameters that may be impaired by cholesterol-lowering drugs, but it cannot constitute a specific test of the hypothesis.
Thus, there is no evidence from clinical trials that can definitively support or falsify the hypothesis that LDL receptor activity governs heart disease risk in the general population, but the current evidence from randomized trials is about as supportive as we might expect assuming the hypothesis is true.Support From Animal Experiments
Given the impossibility of specifically testing the hypothesis in humans, we must turn to experimental evidence in animals and cell culture.
The cholesterol-fed rabbit model is often dismissed by “cholesterol skeptics” because it supposedly does not produce human-like atherosclerosis and because it supposedly cannot be reproduced in animals that are not naturally vegetarians. The first claim is simply false. The second claim is another case of mistaking the diet-heart hypothesis for the lipid hypothesis.
Although the effect of dietary cholesterol is not necessarily generalizable from rabbits to other species, investigators have shown the effect of elevated cholesterol levels in the blood called hypercholesterolemia to be generalizable to many other species including the baboon, cat, chicken, chimpanzee, dog, goat, guinea pig, hamster, monkey, mouse, parrot, pig, pigeon, rabbit, and rat (8). I will cover the collective animal evidence in more detail in a future post.
Anitschkov's drawings of the arterial plaques in his cholesterol-fed rabbit looked very much like those found in human atherosclerosis (9):

Image hosted on the Journal of Lipid Research web site.
{kind=link}
Anitschkov observed first fatty streaks populated by cells from the immune system that we now call “foam cells,” which developed as the disease progressed into a plaque with a necrotic core (a core full of death, or dead cells) containing crystals of cholesterol and calcium deposits, covered by a fibrous cap of smooth muscle cells, just as occurs in humans.Anitschkov and those who reproduced his model were also able to produce a range of severity that mimics what is found in humans. Feeding massive quantities of cholesterol produced a disease state similar to familial hypercholesterolemia, where atherosclerosis develops rapidly and the various internal organs develop deposits of cholesterol. Feeding cholesterol-containing foods such as milk or egg yolks produced a much milder elevation of blood cholesterol levels and atherosclerosis that developed slowly and without abundant cholesterol deposits in the internal organs, as occurs in the general population.Anitschkov also observed that inflammation, high blood pressure, and local arterial injury accelerated this process, while sex hormones, thyroid hormone, and iodine proved protective. He suggested that infection and disturbances of the nervous system were also likely to accelerate the process.
He developed two theories to explain this. The first was his infiltration theory, which corresponds to the lipid hypothesis as defined by Ahrens, Steinberg, and other major figures in the history of the lipid hypothesis. He considered hypercholesterolemia the necessary pre-requisite for human-like atherosclerosis and summarized this view with his dictum that “atherosclerosis never develops without cholesterin.” The second was his combination theory, which held that many other factors determine the final fate of the animal with the disease.
Animal models of atherosclerosis almost never produce a true heart attack, which requires the thinning of the fibrous cap and the release of clotting factors from the lipid-rich necrotic core into the blood, and subsequent formation of a clot that blocks a coronary artery. This may be because rabbits and most other animals synthesize their own vitamin C and are therefore able to continually synthesize collagen even as the inflammatory process degrades it.
In any case, this further supports Anitschkov's combination theory, and suggests that while atherosclerosis may be necessary to produce a true heart attack (of course there are other forms of heart disease and not all are strictly dependent on atherosclerosis), additional factors must also be present. Thus, while elevated levels of cholesterol (in Anitschkov's “infiltrative” view, which I will argue below is wrong) may be necessary to cause atherosclerosis, a multitude of other factors intervene to determine the rate at which atherosclerosis develops and whether it ultimately produces a heart attack.
This is very consistent with the genetic evidence from humans in three important ways.
First, the rabbits seem to develop atherosclerosis when the amount of LDL and related lipoproteins in the blood greatly exceeds the capacity of LDL receptors to clear it. Rabbits with genetically defective LDL receptors develop a very similar presentation (6).
Second, thyroid hormone was able to reverse the disease, and thyroid hormone governs the activity of the LDL receptor. Likewise, dietary cholesterol does not produce atherosclerosis in dogs unless the researchers also inhibit thyroid hormone, suggesting that species that have adapted to eating cholesterol have done so in part by acquiring means of revving up the activity of their LDL receptors.
Third, it is very consistent with the suggestion of human genetic evidence that poor LDL receptor activity is required but not sufficient to produce heart disease. I reviewed evidence above that the PCSK9 mutation increases LDL receptor activity and results in an 88 percent reduction in the risk of heart disease. This suggests, conversely, that the lower LDL receptor activity seen in the general population is virtually required for heart disease to develop.
Alternatively, while those with homozygous familial hypercholesterolemia are essentially guaranteed a very early death, many people with heterozygous familial hypercholesterolemia live to old age without developing heart disease severe enough to be diagnosed. Here's a survival chart from the genetic study I cited above (4):
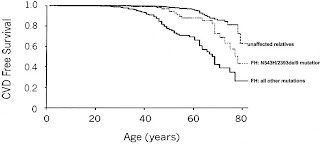
As can be seen above, heart disease begins developing much earlier, around 30, but still by age 40 virtually everyone is still heart disease-free. As the most vulnerable people with familial hypercholesterolemia die off and the people without the disease all start developing atherosclerosis, the slope of the line starts to become similar. By the time these people are in their 70s or 80s, there are far fewer people left without heart disease among those with familial hypercholesterolemia as a result of all of the heart disease that occurred at earlier ages. Nevertheless, even by age 70 or 80, there are people with familial hypercholesterolemia who survive heart disease-free.
Thus, the principle that insufficient LDL receptor activity is necessary but not sufficient to cause heart disease seems generalizable across many species, which makes it very unlikely that humans without specific mutations in the receptor are the sole exception.
So is it true, then, as the proponents of the “lipid hypothesis” suggest, that high cholesterol causes heart disease? No. I believe the evidence suggests that it does not.Cholesterol Does Not Cause Heart Disease
The cholesterol-fed rabbit model provided the striking revelation that high levels of cholesterol in the blood do not cause heart disease.
Here's a slide from my Wise Traditions lecture, Heart Disease and Molecular Degeneration: The New Paradigm, showing how.

If they fed rabbits cholesterol, they developed atherosclerosis. If they injected rabbits with cholesterol, they did not. If they fed rabbits cholesterol, isolated the lipoproteins from their blood that were carrying that cholesterol, and injected those lipoproteins into other rabbits, those other rabbits did develop atherosclerosis.
Thus, it was not the presence of high cholesterol in the blood that caused atherosclerosis, but rather the presence of high amounts of lipoproteins.
Or was it?
Researchers in the late 1970s and early 1980s found that when they incubated LDL particles with endothelial cells, which are the types of cells that make up the inner lining of the blood vessel, something in that LDL changed. It became toxic to those cells. More recent research has continued to confirmed this (10).
Incubating the LDL with those cells for a long period of time approximated the condition of the LDL particle spending a long time in the blood, with the exception that immune cells and many other cell types that would be present in the blood of an organism were absent.
Researchers began calling this “endothelial-cell modified LDL,” but in June of 1984, just three months after Time Magazine hailed the Coronary Primary Prevention Trial (CPPT) as proving that the American Heart Association had been right all along since it first started recommending we replace animal fats with vegetable oils in 1961, Daniel Steinberg's group published a paper in the Proceedings of the National Academy of the Sciences showing that this “modification” was the oxidative destruction of the polyunsaturated fatty acids in the membrane of the LDL particle (11).
This was ironic for several reasons. First, Steinberg was the chair of the CPPT planning committee. Second, polyunsaturated fatty acids are found in the vegetable oils that Time was praising. Third, Steinberg's group had already provided evidence the preceding year suggesting that this oxdiative “modification” of LDL, and not its concentration, promoted heart disease (12):
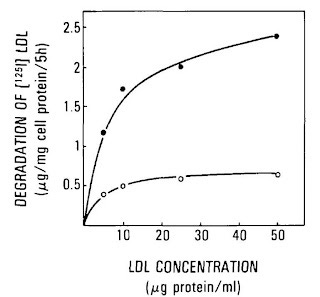
As the line moves upward, immune cells called macrophages have taken up a larger absolute amount of LDL. This is what causes them to morph into the “foam cells” that populate atherosclerotic plaques. As the line moves to the right, the concentration of LDL is increased. The white circles represent fresh LDL and the black circles represent “endothelial-cell modified” (oxidized) LDL.
As we can see, moving from the left side of the graph to the right, the concentration increases five-fold but in the case of the white circles the line stays pretty much flat. This suggests that the concentration of LDL has virtually no effect on the amount that macrophages gobble up. By contrast, oxidation can increase uptake three-fold, five-fold, or more, depending on the concentration.
Later research showed that oxidation occurs in several stages (13). First, polyunsaturated fatty acids in the membrane begin to oxidize. If these products come into contact with the endothelial cells that line the blood, the endothelial cells will produce “adhesion molecules” that attract immune system cells such as monocytes. As the fatty acids continue to oxidize, they eventually destroy the protein in the membrane and prevent its interaction with the LDL receptor, while at the same time facilitating its interaction with the “scavenger receptors” that the monocyte expresses.
Once in the monocyte, the most abundant components of oxidized LDL that are capable of initiating changes in gene expression that turn the monocyte into a macrophage and then into a foam cell are oxidized derivatives of linoleic acid (14), which in the diet are found most abundantly in vegetable oils.
Thus, what we see in this collective evidence is that oxidative destruction of polyunsaturated fatty acids in the membrane of the LDL particle causes it to become toxic to the cells that line blood vessels, and they then send out “help signals” calling on the immune system to clean up the toxic mess, and the immune system then creates an atherosclerotic plaque as an alternative to the greater harm of letting the cells that line the blood vessels die.
Thus, the proximate cause of the development of atherosclerosis is not poor LDL receptor activity, but LDL oxidation. Yet, it is quite clear from the genetic evidence in humans and from animal experiments that insufficient LDL receptor activity is at least one of the major ultimate causes that can lead to LDL oxidation and thus to atherosclerosis.
How? By allowing the LDL particle to spend too long a time in the blood. Unlike the cell, the LDL particle has no vast database of genes nor vast enzymatic machinery to access that genetic information in order to produce antioxidants like glutathione or coenzyme Q10. The liver packages the lipoprotein with a store of antioxidants that will be sufficient if the LDL transports nutrients rather quickly, but if LDL receptor activity is poor, this nutrient transport is compromised, the oxidants within blood deplete the particle of its antioxidants, and oxidation ensues.
The Intrinsic Elusiveness of Biological Truth
Given the complexity of any biological phenomenon, it will be in most cases impossible to
provide a single, definitive demonstration of its truth.
Even the event of LDL oxidation is elusive because cells quickly take oxidized LDL out of the blood in order to protect the blood vessels. There is no specific intervention we can use to increase or decrease LDL receptor activity without having other unintended consequences, whether good or bad. There is likewise nothing we can do to specifically prevent LDL oxidation, since no antioxidants are specific to LDL and since supplementing with single antioxidants can as easily disturb the antioxidant network as it can support it.
Thus, there will certainly be dietary strategies that can minimize LDL oxidation, but none that can do so specifically enough to provide a definitive test of the hypothesis.
I nevertheless believe that the role of lipoproteins in heart disease is one of the most extensively supported biological hypotheses in existence, especially in the realm of human health. The problem is that proponents have so often mistaken this evidence for indicting the concentration of LDL-cholesterol rather than the oxidative degeneration of the LDL membrane that they have misconstrued high LDL-cholesterol as the “cause” or at least a cause of heart disease.
This then brings us to the question of whether we should even pay attention to cholesterol levels or just ignore them.
Do Cholesterol or LDL-Cholesterol Levels Matter?
Although cholesterol levels do not determine our risk for heart disease, they may indeed be a marker for poor LDL receptor activity and related metabolic problems and they should therefore not be ignored.
First, let's dispense with the false notion that cholesterol levels determine our risk for heart disease.
The Framingham data I opened this post with is intriguing, but rather deceptive because of its small sample size, and many people have misused it to suggest that if cholesterol levels are low enough, you simply can't get heart disease.
For example, Caldwell Esselstyn, in his book Prevent and Reverse Heart Disease (see my review here), writes the following:
This is it: if you follow a plant-based nutrition program to reduce your total cholesterol level to below 150 mg/dL and the LDL level to less than 80 mg/dL, you cannot deposit fat and cholesterol into your coronary arteries. Period.
Likwise, in The New Evolution Diet (see my review here), Arthur De Vany refers to an LDL-cholesterol level under 100 as “the no-risk level” as if it is impossible to develop heart disease with cholesterol that low.
Although the Framingham data indicated that everyone with a total cholesterol under 150 mg/dL remained free of heart disease, nine people who underwent screening for the MR FIT trial and had cholesterol levels under 140 mg/dL died of heart disease (15).
Likewise, the single man with a PCSK9 nonsense mutation in the study I cited above who developed heart disease died of a heart attack at the age of 68, but had an LDL-cholesterol level of only 53 mg/dl, well below the supposed “no risk” level. He was obese, smoked, and had high blood pressure. This supports Anitschkov's “combination theory” but flatly contradicts his “infiltration theory,” in which it is the amount of cholesterol rather than the oxidation of
lipoproteins that matters.
Nevertheless, levels of blood lipids, when interpreted in the appropriate context, may be important indicators of metabolic problems, including low LDL receptor activity.
If LDL receptor activity is poor, LDL particles and related lipoproteins will stay in the blood for a longer period of time. The longer they are in the blood, the more they are exposed to cholesterol ester transfer protein, which causes LDL to give triglycerides to HDL in exchange for cholesterol. This will increase LDL-cholesterol and decrease HDL-cholesterol. Furthermore, since cholesterol is more dense than triglycerides, this causes the LDL particles to become small and dense.
At the same time, this causes LDL to continuously encounter oxidants. As its antioxidants become depleted, it begins to oxidize, which makes it even more dense.
The total-to-HDL cholesterol level is a well-established predictor of heart disease risk. This is virtually the same thing as the ratio of LDL-cholesterol to HDL-cholesterol because most cholesterol that is not carried in HDL is carried in LDL.
A recent meta-analysis pooled together the results of 68 prospective studies including over 302,000 participants and found that LDL-cholesterol, HDL-cholesterol, and triglycerides were all capable of predicting the risk of heart disease. When they combined them in a statistical model, however, triglycerides lost their predictive power and only LDL-cholesterol and HDL-cholesterol remained predictive (16):
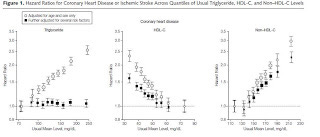
Clicking on the picture will enlarge it. The circles show the unadjusted correlations while the squares show the adjusted correlations.
Likewise, a meta-analysis published in 2007 pooled together the results of 61 studies with almost 900,000 participants (17). These authors were not interested in triglycerides, which allowed them to include some larger studies that didn't measure them. They came to similar conclusions about the predictive power of LDL-cholesterol and HDL-cholesterol, but also concluded that these two factors are independent of one another, so that the total-to-HDL cholesterol ratio provides the most information:
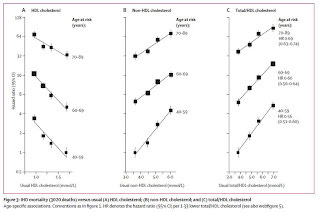
They found that the relative risk of a high ratio decreases with age, but that the absolute risk of a high ratio actually increases with age because so many more people develop heart disease and die from it in old age.
In a future post, I will treat individual studies more thoroughly to examine the nuances. It is quite clear that triglycerides, for example, are not meaningless, because they can be a marker for insulin resistance. Nevertheless, the results of these meta-analyses represent the most consistently powerful blood lipid predictors in the general population.
In a future post, I will also consider other tests that may be useful, such as LDL particle size, emerging tests for oxidized LDL, and others. My point here is simply that the most comprehensively studied, best-established markers do predict heart disease risk and can be seen as markers for poor LDL receptor activity.
Nevertheless, these markers do not determine the risk of heart disease. Therefore, they must be considered clues to look for the real metabolic issues lying behind them.
In the next post in this series, I will consider the possibility that abnormal blood lipids may be markers of poor leptin and thyroid status. I will also take a look at the fact that the natives whose diets we always want to emulate always have “healthy” cholesterol levels.
However, seemingly “bad” changes in blood lipids could also indicate positive healing processes. As I pointed out in a post recently, “Why Is My Cholesterol So High On This Diet?” an increase in cholesterol or triglycerides following a dietary change could indicate that the diet is curing fatty liver, or that the cells are faced with a new abundance of energy that they can use for productive processes, both of which are good things. They may also accompany weight loss, which emphasizes the need to measure blood lipids after a period of stable diet and weight. And finally, a number of factors may increase cholesterol synthesis or decrease the oxidation of VLDL and LDL particles either before they are sent from the liver or once they are in the blood, both of which could raise blood lipids.
Thus, these markers, in my view, should neither be ignored nor interpreted out of context.
Interpreting that context may be difficult and nuanced, and I will consider these nuances in future posts in this series.
References
1. Kannel WB, Castelli WP, Gordon T. Cholesterol in the prediction of atherosclerotic disease. New Perspectives Based on the Framingham Study. Ann Intern Med. 1979;90(1):85-91.
2. Rose V, Wilson G, Steiner G. Familial hypercholesterolemia: Report of coronary death at age 3 in a homozygous child and prenatal diagnosis in a heterozygous sibling. J Pediatr. 1982;100(5):757-9
3. Fredrickson DS, Levy RI. Familial Hyperlipoproteinemia. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, eds. The Metabolic Basis of Inherited Disease. New York, NY: McGraw-Hill (1972). pp. 531-544.
4. Umans-Eckenhausen MA, Sijbrands EJ, Kastelein JJ, Defesche JC. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation. 2002;106(24);3031-6.
5. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-72.
6. As reviewed in Goldstein JL, Kita T, Brown MS. Defective lipoprotein receptors and atherosclerosis. Lessons from an animal counterpart of familial hypercholesterolemia. N Engl J Med. 1983;309(5):288-96.7. Taylor F, Ward K, Moore TH, Burke M, Davey Smith G, Casas JP, Ebrahim S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2011;19(1):CD004816.
8. Steinberg D. The Cholesterol Wars: The Skeptics vs. the Preponderance of the Evidence. San Diego, CA: Academic Press (2007).
9. Anitschkow N. Experimental Arteriosclerosis in Animals. In: Cowdry EV. Arteriosclerosis: A Survey of the Problem. New York: Macmillan (1933).
10. Sata M, Walsh K. Endothelial Cell Apoptosis Induced by Oxidized LDL is Associated with the Down-regulation of the Cellular Caspase Inhibitor FLIP. J Biol Cehm. 1998;273(50):3303-6.
11. Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci USA. 1984;81(12):3883-7.
12. Henrisksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of biologically modified low density lipoprotein. Arterioscelrosis. 1983;3(2):149-59
13. Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witzum JL, et al. Structural Identification by Mass Spectrometry of Oxidized Phospholipids in Minimally Oxidized Low Density Lipoprotein That Induce Monocyte/Endothelial Interactions and Evidence for Their Presence in Vivo. J Biol Chem. 1997;272(21):13597-13607.
14. Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93(2):229-40.
15. Iso H, Jacobs DR, Wentworth D, Neaton JD, Cohen JD. Serum cholesterol levels and six-year mortality from stroke in 350,977 men screened for the multiple risk factor intervention trial. New Engl J Med. 1989 Apr 6;320(14):904-10.
16. Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009;302(18):1993-2000.
17. Prospective Studies Collaboration, Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. Blood cholesterol and vascular mortality by age-sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370(9602):1829-39.